本文內(nèi)容僅可為解決實(shí)際問題時(shí)提供一個(gè)參考,不作為2010版GMP的依據(jù)或判定原則。
1、針劑在線滅菌時(shí):
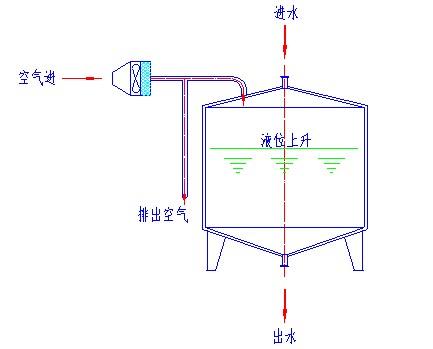
A)呼吸過濾器如何保證達(dá)到無菌保證要求?
B)濾芯在滅菌浸濕后,通氣效果變差,如何處理有效?
解答:
呼吸器過濾器是可以作SIP或離線滅菌的,它的要求取決所使用的目的。如純化水貯罐上用的呼吸器過濾器可采用離線滅菌法(沒有必要設(shè)在線滅菌),也可采用下圖所示高效過濾器主動(dòng)送風(fēng)的形式,在任何情況下保持貯罐的正壓狀態(tài)。
凍干及其它無菌生產(chǎn)設(shè)備上的疏水性過濾器的滅菌,通常采用電加熱或壓縮空氣吹干的方法使其保持運(yùn)行狀態(tài),但是不同的情況有不同的處理方式,具體可以按供貨商規(guī)定的要求處理。
2、壓縮空氣系統(tǒng)確認(rèn)中測定壓縮空氣中水份的含量(使用德爾格壓空儀測定)水份含量指標(biāo)換算后即可代表露點(diǎn)值?該含量可接受標(biāo)準(zhǔn)應(yīng)如何考慮,是否不同潔凈級別之間水份要求不同?無油壓縮機(jī)是否可不測試含油量?
解答:
壓縮空氣的控制標(biāo)準(zhǔn)是結(jié)合所需生產(chǎn)的藥品特征而設(shè)計(jì)控制標(biāo)準(zhǔn)的,有露點(diǎn)、微粒、含油量、微生物限度等。壓縮與生產(chǎn)區(qū)的潔凈級別沒有多大直接相關(guān)性。壓縮空氣有國標(biāo),企業(yè)應(yīng)根據(jù)產(chǎn)品情況制訂本企業(yè)的標(biāo)準(zhǔn),原則上說,它不得影響產(chǎn)品的工藝及質(zhì)量。
因?yàn)楝F(xiàn)空氣的污染情況比較嚴(yán)重,無油空氣壓縮機(jī)并不可能解決“空氣污染問題",采用無油空氣壓縮機(jī)時(shí)仍應(yīng)對含油量控制。
3、壓縮空氣應(yīng)確認(rèn)哪些項(xiàng)目?現(xiàn)場檢查中要檢哪些指標(biāo)?如何檢查?
解答:
在制藥企業(yè)中,壓縮空氣常見的二種用處分別是:氣動(dòng)閥的動(dòng)力源;生產(chǎn)工藝用氣體,例如洗瓶的吹干、滅菌柜的補(bǔ)氣(特別是滅菌時(shí)作軟包裝的背壓)以及配制罐壓藥液等等。動(dòng)力源通常只有壓力的要求,但工藝用氣體要考慮產(chǎn)品的特點(diǎn),如是無菌分裝,對干燥的要求可能會高一點(diǎn)(控制露點(diǎn));如是液體產(chǎn)品,則不需要特別干燥,因此,GMP規(guī)范不規(guī)定技術(shù)要求,檢查方法請見國標(biāo)。注意,不要離開了產(chǎn)品的工藝要求,去提露點(diǎn)(干燥度)、微粒等限度標(biāo)準(zhǔn)。
4、過濾系統(tǒng)終端過濾芯的完整性測試試驗(yàn)一定需要在線監(jiān)測嗎?而壓縮空氣系統(tǒng)、制氮系統(tǒng)的濾芯如何測試和清潔?
解答:
GMP并沒對此規(guī)定,有條件時(shí)建議在線監(jiān)測,但這不是強(qiáng)制要求。與液體除菌過濾器不同,壓縮空氣系統(tǒng)、制氮系統(tǒng)除菌過濾器的濾芯可按供貨商提供的方法進(jìn)行測試,定期檢查;其前道其它用處的過濾器(例如去除油霧),均應(yīng)按工藝要求及供貨商要求處理。
5、大輸液生產(chǎn)過程中罐裝及管路的批次清場有何要求?批與批之間必須在線清洗嗎?
解答:
這是一個(gè)比較現(xiàn)實(shí)的問題,清洗的目的是什么,要解決什么問題?因?yàn)榍逑春螅A罐及管路等會有部分積水,除非你用壓縮空氣或氮去吹干,否則會影響下批初始產(chǎn)品的濃度,清洗需要操作時(shí)間,也會造成浪費(fèi)。因此,如是批與批(同一產(chǎn)品、相同濃度及包裝規(guī)格)的連續(xù)生產(chǎn),可能不作清洗,也可能只需要簡單的清洗(只清洗貯罐),這要看產(chǎn)品是否容易分解、長菌或其它對產(chǎn)品質(zhì)量可能的不良后果。換言之,應(yīng)根據(jù)風(fēng)險(xiǎn)評估的結(jié)果作出決定。

6、滅菌冷卻過程中為了維持瓶內(nèi)外壓力平衡,需補(bǔ)充壓縮空氣,請問此使用點(diǎn)的壓縮空氣是否需要除菌過濾?
解答:為了避免已滅菌產(chǎn)品被再次污染,補(bǔ)充的壓縮空氣應(yīng)經(jīng)除菌過濾。
7、用于塑料瓶氣洗的終端壓縮空氣濾器完整性實(shí)驗(yàn)如何做?怎樣才能有好的辦法檢測到濾芯漏了?
解答:
新版GMP附錄1無菌藥品第四十二條進(jìn)入無菌生產(chǎn)區(qū)的生產(chǎn)用氣體(如壓縮空氣、氮?dú)猓话扇夹詺怏w)均應(yīng)經(jīng)過除菌過濾,應(yīng)當(dāng)定期檢查除菌過濾器和呼吸過濾器的完整性。請注意,氣體過濾器的截留效率比液體過濾器高出10~100倍。國際上,將氣體過濾器分為3類:
◇ 直接與無菌產(chǎn)品接觸(如:無菌灌裝中使用的壓縮空氣)
◇ 不直接與無菌產(chǎn)品接觸(如:發(fā)酵過程通氣過濾器)
◇ 降低微生物負(fù)荷(微生物污染水平)(如:HVAC中的HEPA高效過濾器)
所提塑料瓶是用于無菌藥品還是非無菌藥品生產(chǎn)不明確,這里分別說明一下。如果是無菌生產(chǎn)(如凍干),凍干腔室和工藝用氣體除菌過濾器是做在線滅菌的,但通常不是每批進(jìn)行在線滅菌,其完整性也不每批檢測,而是定期測試,這樣做的風(fēng)險(xiǎn)是--如后一次測試證明除菌過濾器確實(shí)損壞,則從上次測試合格后生產(chǎn)到這次測試期間的產(chǎn)品將作無菌不合格處理。為了避免經(jīng)濟(jì)的風(fēng)險(xiǎn),可購買在線完整性測試儀進(jìn)行測試;如是最終滅菌產(chǎn)品,過濾器實(shí)際上可長期使用,定期檢查,例如這種檢查可以是每半年、一年檢查一次,由企業(yè)根據(jù)實(shí)際使用情況及結(jié)果進(jìn)行風(fēng)險(xiǎn)分析,確定檢查周期。
8、公用系統(tǒng)(如壓縮空氣系統(tǒng)、制氮系統(tǒng))其管道是否需要定期消毒?如何做?
解答:
GMP對此無技術(shù)性規(guī)定,應(yīng)按供貨商的維修要求處理,管路的清潔應(yīng)在安裝階段完成。用作動(dòng)力源的壓縮空氣不需要消毒。工藝用壓縮空氣系統(tǒng)、制氮系統(tǒng)的管路,用于最終滅菌產(chǎn)品時(shí),也不要求消毒;當(dāng)于不可最終滅菌的產(chǎn)品(已除菌過濾后的藥液)接觸時(shí),部分管路(軟管)與過濾器是經(jīng)滅菌的。
9、與藥液直接接觸的壓縮空氣或氮?dú)庠谙到y(tǒng)安裝時(shí)不進(jìn)行全面的驗(yàn)證,日常監(jiān)測是否可只監(jiān)測含菌量及粒子?
解答:
公用介質(zhì)通常采用確認(rèn),而不稱驗(yàn)證。與藥液直接接觸的介質(zhì)系統(tǒng)均應(yīng)確認(rèn),未經(jīng)確認(rèn)的系統(tǒng)不應(yīng)投入使用。值得強(qiáng)調(diào)的是,含油量是常見的確認(rèn)項(xiàng)目,應(yīng)列入確認(rèn)方案,并在確認(rèn)中進(jìn)行確認(rèn)。
壓縮空氣和氮均有國家及ISO/國際標(biāo)準(zhǔn),網(wǎng)上可查得到標(biāo)準(zhǔn)的具體要求。不同產(chǎn)品對這類介質(zhì)的要求不盡相同,因此,標(biāo)準(zhǔn)由企業(yè)根據(jù)產(chǎn)品來定,GMP對這類介質(zhì)無技術(shù)性規(guī)定。另外,用于無菌生產(chǎn)(例如凍干粉針)的壓縮空氣或氮,均應(yīng)通過除菌過濾。
10、工藝用壓縮空氣管道系統(tǒng)定期消毒的可操作性?能否理解:選用的無油空壓--經(jīng)過干燥--三級過濾--確認(rèn)后在和物料接觸前端再加0.2um過濾后,管道系統(tǒng)定期消毒,只要對壓縮空氣品質(zhì)監(jiān)測評估即可?
解答:
所提到的處理方法基本都是這樣,但要根據(jù)風(fēng)險(xiǎn)來討論要求。經(jīng)處理的壓縮空氣進(jìn)生產(chǎn)區(qū)時(shí),可能有二種情況,一種是用作動(dòng)力源(平時(shí)不列入監(jiān)控范圍);另一種是工藝或動(dòng)力二者的結(jié)合(因簡化管路而不將管路分得過細(xì))。只有無菌藥品的生產(chǎn)中,在分管路上才安裝除菌過濾器,保證直接與藥品或內(nèi)包材接觸的氣體符合工藝要求,需要做的是定期檢查除菌過濾器的完好性。工藝用壓縮空氣要檢查含油量和微生物項(xiàng)目。
用于無菌生產(chǎn)(全無菌操作)的壓縮空氣,如凍干機(jī)平衡壓力用的壓縮空氣,或用于壓無菌藥液的壓縮空氣等,過濾器與容器往往是做在線滅菌的,或部分軟管與過濾器一起滅菌。




 咨詢電話
咨詢電話